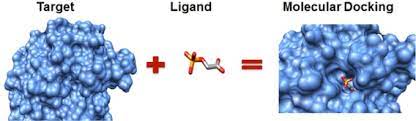[강화학습 스터디] Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation + SAC, PER
로몽25
2022. 6. 3. 17:27
Intro
이번 논문의 리뷰를 남기게 된 계기는, 지난 3개월 간의 배움의 과정을 집약적으로 설명하기 위함이기에 다소 주관적이고 논문 한 건의 리뷰 이상을 담고자 했음을 밝힌다. 해당 논문을 제대로 이해하기 위해서는 SAC(soft actor critic), PER(Prioritized Experience Replay)와 같은 비교적 최신의 강화학습 모델 알고리즘뿐 아니라, CADD라는 컴퓨터 기반 신약개발 분야의 도메인 지식과 특징의 이해도 필요하므로, 각각을 Domain Background, Tech Background로 나누어 서술한 후 본격적인 논문의 모델 리뷰로 들어가기로 했다.
Domain Background
1. CADD (Computer-aided Drug Design)
: CADD란 신약 개발 과정에 활용되고 있는 컴퓨터 기반 신약 디자인 과정으로, 화학물의 입체 구조 예측, 의약품 활성, 독성 예측 등 다양한 단계에 접목되고 있는 융합 학문적 기술이다.
위 그림에서 보면, 각 단계 별로 수행해야 할 task들이 매우 다양한 것을 알 수 있다. 이젠 살짝 오래됐다고 느껴질 법도 한 DeepMind의 Alpha Fold(2018,https://alphafold.ebi.ac.uk/)는 저 단계 중에서도 초반부의 Target Identification : Protein Structure Prediction에 해당하는 기술이라고 볼 수 있다. 이번에 주목할 부분은 Lead Discovery : Docking scoring, De novo design, Pharmacophore 단계이다.
2. Docking
: Docking(도킹)이란, 비교적 큰 크기의 protein(단백질, 혹은 receptor라고도 한다)와 작은 크기의 ligand(리간드, 분자량도 작고 크기도 작고 구조도 간단한 조각을 지칭한다) 사이에서 일어나는 결합 과정을 의미한다. 보통 하나의 단백질이라고 해도 여러 군데의 active site(활성 부위, 결합이 일어나는 장소)를 가지고 있는데, 어떤 리간드가 제시되느냐에 따라 활성 부위가 달라질 수도 있고, 미처 예상하지 못했던 부분이 활성 부위가 될 수도 있고, 어떤 강도(binding affinity)로 결합하는지도 달라지기 때문에 docking 과정을 예측하는 것은 상당히 복잡하고 어렵다.
불행인지 다행인지, docking의 결합력을 예측해서 docking score를 내는 연구와 프로그램은 상당 수 분자 화학에 기반해서 진행되었고, 실제로 많은 연구에서 활용될 만큼 나름 괜찮은 성능을 보여준다. 그중 가장 유명하고 이름 높은 프로그램은 AutoDock Vina(https://vina.scripps.edu/)인데, 분자 화학에 기반한 에너지 모델을 사용한 분자 모델링 시뮬레이션 프로그램이다. 프로그램으로 배포되기도 하고, code 형태로 커맨드 창에서 돌릴 수 있게 라이브러리 형태로 제공되기도 한다. (무엇보다 가장 큰 장점은 무료라는 것이다.) AutoDock Vina와 그 변종에 대한 자세한 정보는 링크를 참고하길 바란다.
다만 사용시 주의할 점은, input file로 넣어주는 pdbqt 형태가 일반적인 단백질 정보 파일(.pdb)과 달라 변환 과정에서 오류가 생기기 쉽다는 것이다.
AutoDock Vina는 이후 Vina GPU 버전, qvina, smina, GNINA 등 다양한 변종이 만들어졌지만, 해당 프로젝트를 진행하면서 내가 겪어본 바로는 vina가 benchmark로 가장 적당하고, qvina가 빨라서 많은 수의 ligand를 훑어볼 때 유리한 것 같다.
AutoDock Vina처럼 에너지 모델링에 기반한 프로그램도 있지만, 최근에는 딥러닝의 일반화 & 활용 트렌드에 힘입어 Docking 모델을 딥러닝으로 해결하려는 시도들도 많이 생겼다. 모델의 형태에 기반해 몇 가지를 소개하자면,
이 외에도 정말 다양한 딥러닝 모델들이 제시되고 있으며, 원자와 결합으로 구성된 분자(protein, ligand)의 형태와 비슷한 graph network에 기반한 연구가 많이 진행되고 있는 것으로 보인다. graph network가 computer vision이나 NLP에서 대중적으로 많이 쓰이는 모델이 아닌 까닭에 연구 발전 속도에 있어 아직은 가속화될 여지가 많이 남아있다고 판단된다.
3. Hits & De novo design
: 그렇다면 논문 제목의 일부인 'Hits'에 대해 이해해보자.
신약 개발, 더 좁히면 해당 타겟에 대해 '유효한' ligand를 찾는 과정에서 'hit molecule'이란
Molecules with high therapeutic potential
High binding affinity to a given protein target
의 조건을 만족하는 분자라고 할 수 있다. 다시 말해, docking 결과가 좋고 실제로 의약품 활성을 나타낼 수 있는 분자(=리간드)라고 할 수 있고, 전체 분자들 중에서 이러한 'hit molecule'을 얼마나 잘 찾아내는가가 바로De novo design단계의 목표라고 할 수 있다.
전체 분자들을 각각 하나의 sample로 취급한다면 이 분자들이 있는 공간을 searching space라고 할 수 있다. 이때, 우리는 random 하게 searching space를 돌아다니면서 분자들을 테스트(docking, toxicity, effectiveness 등을 검사)할 수도 있지만, 시간, 비용 모두에 있어 비효율적일뿐더러 실제 실험이 아니라 컴퓨팅 파워를 활용하는 의미가 퇴색될 수밖에 없다.
그렇다면, searching space에서 적합한 path를 탐색하도록 강화학습을 통해 agent를 학습시킨다면 훨씬 효율적으로 새로운 분자들을 탐색할 수 있지 않을까?
강화 학습을 사용한다면, 두 가지를 검토할 필요가 생긴다. 첫 번째는 강화 학습 모델 알고리즘이고, 다른 하나는 reward이다. 특히 reward는, 어떤 모델 알고리즘을 쓰더라도 결론적으로 도출하는 분자가 실제 'hit molecule'로 이어지기 위해서 중요한 요인이 된다. 탐색 공간에서 새로 찾아낸 분자에 대해 실험 장비를 통해 직접 docking 등을 계산할 수는 없기 때문에, reward를 계산하는 것도 시뮬레이션 모델을 쓸 수밖에 없고, 그 점에서 uncertainty를 한 단계 더 증가할 수밖에 없다. 따라서 강화 학습 agent를 사용하게 된다면, 두 가지의 task가 발생한다.
Task 1. searching space 내에서 더 다양한 분자들을 찾을 수 있도록 하는 (more explorative) 모델 알고리즘 Task 2. 더 정확한 reward function/model (more accurate reward)
위의 두 가지 task를 얼마나 잘 해결하는지가 CADD의 첫 단계를 얼마나 잘 통과할지 여부를 결정한다고 해도 무방하지 않을까, 하는 의견이다.
Technical Background (RL)
1. SAC : Soft Actor Critic
https://sites.google.com/view/soft-actor-critic
: Soft Actor-Critic: Off-Policy Maximum Entropy Deep Reinforcement Learning with a Stochastic Actor (ICML 2018) 논문에서 소개되었다. 간단하게 설명하자면 actor는 searching space(탐색 공간)을 돌아다니면서 expected reward를 최대화하는 동시에 exploration entropy도 최대화 하는 방향으로 행동을 수행하도록 짜인 모델 알고리즘이다. 다시 말해, 랜덤 하게 행동하면서 보상의 최대화를 꾀한다는 것이다.FREED를 이해하는데 있어 SAC를 A to Z로 알고 있을 필요가 있을까 싶기는 했지만, RL를 하는데 SAC를 모른다는 건 트렌드에 뒤떨어진 자가 되는 것이란 생각이 들어서 추가로 공부했다.
RL을 사용할 때 탐색할 환경에 대해 잘 모르는 경우, 우리는 대부분 model free Reinforcement Learning 방식을 사용한다. 게임뿐 아니라 real world의 다양한 문제들은 복잡할뿐더러 환경이나 reward 등 밝혀진 바가 없기 때문에 model-free deeplearning RL을 사용하게 된다. 하지만 이런 방식을 사용하면 고전적으로 두 가지 문제점이 발생할 수밖에 없다.
높은 샘플 복잡도: sample space가 복잡하거나 sample 자체의 표현이 복잡한 경우가 많다.
하이퍼 파라미터 수정 의존성이 높은 것: 하이퍼 파라미터를 어떻게 설정하는가에 따라 모델의 성능이 크게 바뀌는 현상으로, 모델의 성능을 단순 예측하기 어렵고 여러번 섬세한 조정이 필요하다는 점에서 어려움이 생긴다.
따라서 샘플 효율성을 늘리기 위해 매 action마다 새로운 data sample을 사용해서 학습에 사용하는 것 대신, 과거의 경험을 누적해서 사용하는 방식을 사용하면서 보다 일반화되고 안정적인 모델 알고리즘을 만들기 위해,최대 엔트로피 프레임 워크를 사용하여 엔트로피 최대화 기간으로 표준 최대 보상 강화 학습 목표를 확장한 soft actor critic을 제안했고, 해당 모델은 여러 검증을 거쳐 높은 샘플 효율성과 안정성, 그리고 exploration 향상을 보였다.
Google DeepMind에서 2015년 낸 논문으로, 기존 DQN(Deep Q Network)에서 사용하던 Experience Replay 테크닉을 개선한 방식이라고 할 수 있다. 기존의Replay Buffer은 경험의중요성과 상관 없이 buffer에 있는 과거 sample들 중에서 random 하게 뽑아서 학습을 시켰는데, 거기에중요성을 기반으로 우선 순위를 부여하여 학습에 사용되도록 한 방식이라고 할 수 있다.
Q. Q-learning와 DQN Q-learning을 매우 직관적으로 설명한 위키피디아 설명. 느낌만 참고하면 좋을 것 같다.
: Q-learning, Q function, Q value 모두 같은 개념을 가리키는 말에서 시작한다. 이때 Q는 quality를 의미하는데, Q value는 따라서 보상(reward)과는 전혀 다른 개념임을 가장 먼저 인지해야 한다. Q value란, 현재 상태에서 특정 action을 취할 때, 미래의 기대 보상을 의미한다.
따라서 Q-learning에서 Q function을 제대로 학습시키기 위해 계속적으로 sample을 이용하는 점을 기반으로, Q function을 더 복잡한 real world 문제에서도 작동하도록 하기 위해 근사된 deep network로 변경한 것이 DQN의 주된 아이디어라고 할 수 있다.
하지만 단순히 Deep Network로 Q function을 치환한다고 해서 모델이 잘 돌아가지는 않았고, 두 가지 문제점이 발생했다.
1) 딥러닝은 label이 있는 데이터를 학습시키는 데 치중하고 있는데, 강화학습은 label이 존재할 수 있는 구조가 아니고, 띄엄띄엄 들어오는 reward를 통해서 학습시켜야 했다. 2) 딥러닝에선 각 sample들이 독립적이라는 (i.i.d) 가정에 기반하지만, 강화 학습의 각 sample들은 현재 state와 다음 state 등의 형태로 이루어져 있어 연관성이 크기 때문에 해당 가정이 성립하지 않는다.
DQN DQN에서 도입한 Experience Replay는, 강화학습 episode를 진행하면서, 각 step마다 deep network의 가중치를 업데이트하기보단, [S(Current State), A(Action), R (Reward), S'(Next State)]의 데이터셋을 buffer에 저장해 두고 episode가 종결될 때 random 하게 뽑아서 학습시키자는 의도였다. reward가 드물게 들어오는 1번 문제점을 해결하는 동시에, random하게 sample을 뽑아서 학습 셋에 제공함으로써 각 sample 간의 연관성이 감소하게 되어 2번 문제점을 해결할 수 있었고, 과거의 경험을 계속 buffer에 가지고 있음으로써 샘플 효율성을 향상하는 효과까지 볼 수 있었다.
그렇다면 다시 돌아와서, PER(prioritized Experience Replay)로 돌아오자. buffer에 저장된 sample을 우선순위를 정해 뽑아서 사용함으로서, PER은 새로운 효과를 볼 수 있다. 중요한 경험(experience, sample)을 더 많이 학습하는 데 사용할 수 있다는 것이다. 경험의중요성은,현재의 Q Network를 이용해 기존의 경험과의 TD-error를 구한 후 buffer에 함께 저장된다.
PER 알고리즘
하지만, 우선순위를 가지고 replay할 sample을 뽑아오는 형태이므로 우선순위를 정하는 metric을 어떤 것을 정하는지에 따라 성능이 달라질 수 있다. PER 논문에서처럼 일반적으로 TD error(predictive error)를 많이 쓰지만, 이제 소개하게 될 FREED에서는 Bayesian Uncertainty(일종의 variance)를 metric으로 사용하여 TD error와 비교했다. (PER(BU), PER(TD))
Paper & Model Review
1. Domain method
1) fragment-based molecular generation
: 단순히 다양한 분자들을 마구잡이로 가져오는 것이 아니라, molecule library에 이미 넣어두었던 fragment를 차례대로 하나씩 붙이는 방식을 사용했고, 따라서 분자 구조적으로 말이 되지 않는 ligand는 배제하는 동시에 전체 protein의 활성 부위 안에 결합할 수 있는 ligand로 한정할 수 있도록 했다.
: 1step은 서로 다른 의미를 가진 총 3개의 action으로 구성되어 있는데,
action 1) ligand의 각 원자들 중에서 새로 fragment가 붙을 수 있는 위치를 찾는 action (possible attachment site)
action 2) 실제로 붙을 수 있는 fragment 선택하는 action
action 3) 기존의 ligand의 부착 위치(action1)와 새로 선택된 fragment(action2) 사이의 결합 원자, 각도 결정
1 step을 통해 action 1,2,3이 모두 수행되고 나면 기존의 ligand는 다음 state인 S(t+1)로 변하게 된다.
2) reward
: reward는 매 step마다 주지 않고, 정해진 step(배포된 모델의 경우 25 step마다)docking program(배포된 모델의 경우 qvina, 다른 프로그램 사용 가능)을 사용해서 reward를 제공했다. 나머지 step에 대해선 reward predictor network를 따로 두어 각 step을 통해 생성된 새로운 분자들의 reward를 예측하는 데 사용했다. 25 step마다 docking simulation을 돌리면서, 동시에 reward predictor 네트워크를 업데이트하는 데 사용하고 이때 발생하는 predictive error를 따로 저장하여 buffer에 함께 저장하여 우선순위로 사용하기도 했다. (metric = Predictive Error인 경우)
2. RL method
: SAC(soft actor critic) + PER(Prioritized Experience Replay)
기존의 분자 생성 모델들의 경우, 결론적으로 비슷한 분자들만 생성하는 등, searching space를 제대로 탐색하지 못하는 한계점이 있었고, 이 논문에선 그러한 한계점을 해결하기 위해 최대한 exploration를 보완하기 위한 method를 사용했다. 먼저 SAC를 활용하여 탐색 과정에서 최대 엔트로피를 추구하는 동시에, PER 테크닉을 함께 사용했다.
1) PER
위에서 소개했던 original PER의 목표가 샘플 효율적인 탐색을 위한 것이었다면, 해당 논문에서 사용한 이유는 '다양성'을 위해서였기 때문에 metric을 변형해서 사용했다. 선택된 metric은 PE(predictive error), BU(Bayesian uncertainty)으로, 특히 BU는 일종의 variance로 sample의 새로움을 대변하는 metric이다.
2) SAC
: PER을 적용하면서 살짝 변형됐지만 전체적인 골자는 SAC을 따르고 있다. PER를 통해 특정 step마다 우선순위에 따라 뽑히는 sample을 actor-critic network를 학습시키는 데 사용하고, 다시 해당 과정을 반복하는 형태로 이루어진다.
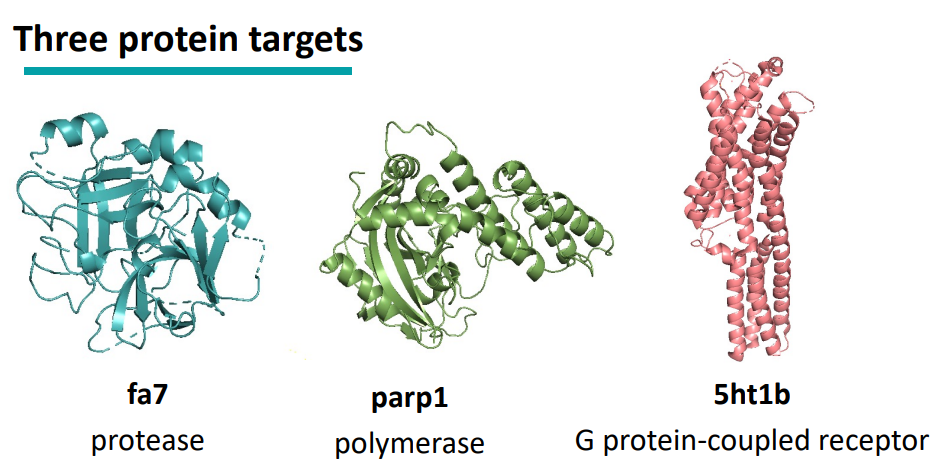
3. Evaluation
: 3개의 예시 protein에 대해 docking을 기반으로 molecule들을 생성하여, evaluation metric에 따라 평가했다.
Quality score (filter score) : 기존에 존재하는 특정 filter(조건식)을 사용하여, [유효한 분자 개수/생성된 전체 분자 개수] 계산해서 구한다. 이때 유효하다는 것은 filter의 조건들을 모두 통과했음을 의미한다.(ex filters = Glaxo, SureCHEMBL, PAINS)
Hit ratio : [active한 분자들의 docking score 중간값보다 높은 값을 갖는 분자들의 개수/생성된 전체 분자 개수]
Top 5% score : [docking score top 5%의 평균값]
출처 : 논문 원문실제 github 코드 돌려본 이미지 : Hit ratio, Top 5% score 확인 가능
Conclusion
FREED 모델은 복잡한 real world 문제 중 하나인 'CADD : fragment-based molecule generation'에 대해 도메인 아이디어와 exploration을 강조한 RL 모델을 함께 융합하여 진행한 연구의 결과라고 할 수 있다. 하지만 여전히 이 모델조차도 완벽한 분자 생성 RL 모델이라고 볼 수는 없다. 더군다나, 해당 모델은 reward로 docking만을 제공하기 때문에 의약품으로써의 다른 특성(ADMET)을 검증할 수 없다는 점에서 CADD의 해당 단계를 온전히 대체할 수 없다는 한계가 있다. 그럼에도 불구하고 최근의 CADD RL 모델 중에서 가장 domain과 RL 테크닉을 잘 융합한 모델 중 하나가 아니었을까 하는 생각을 해본다.